|
||||||||||||
 |
 |
 |
 |
 |
 |
 |
 |
| 购买进口仪器、试剂和耗材——就在始于2001年的毕特博生物 www.bitebo.com |
免责声明:本文内容来自互联网,如有不慎侵害的您的权益,请告知我们,将尽快删除。
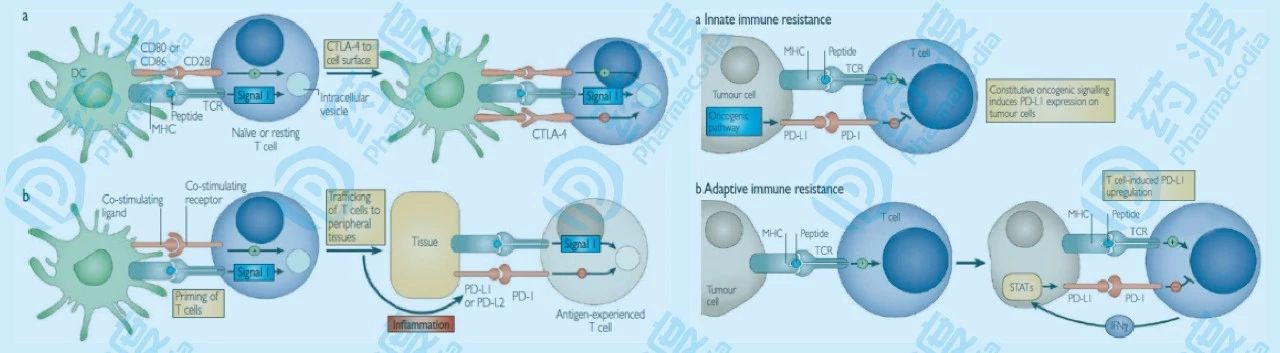
1. 肿瘤细胞正在“胜利大逃亡” 去年备受关注的诺贝尔生理学或医学奖,让“免疫负调控”的发现者詹姆斯·艾利森和本庶佑声名显赫。“免疫负调控”发生在抗原呈递期间,即初始T细胞与抗原呈递细胞之间传递抗原信号,以及细胞识别期间,即效应T细胞迁移进入肿瘤组织,与肿瘤细胞或免疫细胞之间传递识别信号期间。 大部分肿瘤细胞就是充分利用“免疫负调控”,来抑制细胞毒性T细胞的免疫活性,从而逃避免疫系统的追杀。具体地说,在正常状态下,当炎症反应发生时,NK细胞、T细胞、巨噬细胞、树突状细胞等免疫细胞,以及表皮细胞和血管内皮细胞表面会被诱导表达PD-L1蛋白。当这些细胞和被激活的T细胞接触时,PD-L1与T细胞表面的PD-1结合,从而抑制T细胞的免疫活性,避免过激的炎症反应对自身的伤害。因为这些细胞表面表达的PD-L1程度比较低,所以可以避免对T细胞活性的消耗。然而肿瘤细胞大不一样,它们在细胞表面大量表达PD-L1,能够几乎完全抑制与它们接触的所有T细胞的免疫活性,造成T细胞活性耗竭,并逃避免疫系统的追杀,最终恶性繁殖扩增,危及生命。(图1)
图1 :免疫负调控示意图 (a)当T细胞对抗原产生初次响应时,CTLA-4介导的免疫检查点被诱导激活。这种由CTLA-4介导的诱导激活程度,依赖于起始T细胞受体调整的信号强度。高亲和配体能够诱导表达更多的CTLA-4,从而减弱了起始响应的强度。当T细胞受体遭遇抗原后,诱导下游通路,CTLA-4被转运到细胞表面,此时CTLA-4起到信号减弱的功能,以维持一个恒定的T细胞激活水平。(b)与CTLA-4不同,PD-1信号通路并不在起始T细胞激活阶段起作用,而是在外周组织中,效应T细胞识别组织中的抗原,以调节炎症响应的过程中。这些组织中的炎症信号(IFN-γ,主要由I型辅助T细胞表达)能够诱导组织细胞中的PD-L1的表达,从而抑制效应T细胞对其免疫响应。在慢性抗原暴露的情况下,T细胞表面过量诱导的PD-1,可以引发T细胞的活性耗竭。(c)在肿瘤细胞中,PD-L1的表达或不依赖于肿瘤微环境中的炎症信号,AKT、STAT3信号通路的激活可诱导表达PD-L1,或依赖于炎症信号,表达下游的免疫检查点抑制蛋白。
目前,肿瘤的联合免疫疗法越来越体现出它的优越性。一方面,要减少肿瘤靶向结合的非特异性,减少治疗过程对正常细胞的杀伤,我们需要尽可能地将免疫系统引起的细胞毒性局限在肿瘤组织中(靶向药物)。另一方面我们需要解除肿瘤细胞和免疫细胞之间的免疫负调控,提高免疫细胞对肿瘤细胞的细胞毒性(免疫负调控抑制),使肿瘤杀伤单抗药物的肿瘤杀伤作用更能发挥威力。 在这些治疗方案中,都以T细胞免疫为核心。虽然细胞毒性T细胞的肿瘤杀伤作用具有一定的特异性,然而极优而劣。因为细胞毒性T细胞的特异性,来源于被杀伤细胞的MHC-I型抗原递呈。只有识别了目标细胞通过MHC-I递呈的抗原,细胞毒性T细胞才能特异性的杀伤靶细胞。然而,狡猾的肿瘤细胞,有相当一部分关闭了其细胞表面MHC-I类分子的表达,比如,92%的宫颈癌细胞,71%的乳腺癌细胞,64%的非小细胞肺癌细胞。这样我们英勇无比的细胞毒性T细胞,对它们就无能为力了。 “如之奈何?” 幸运的是,对付这类狡猾的肿瘤细胞,我们有免疫系统的另一杀手,自然杀伤细胞(Natural Killer Cell, NK细胞)。
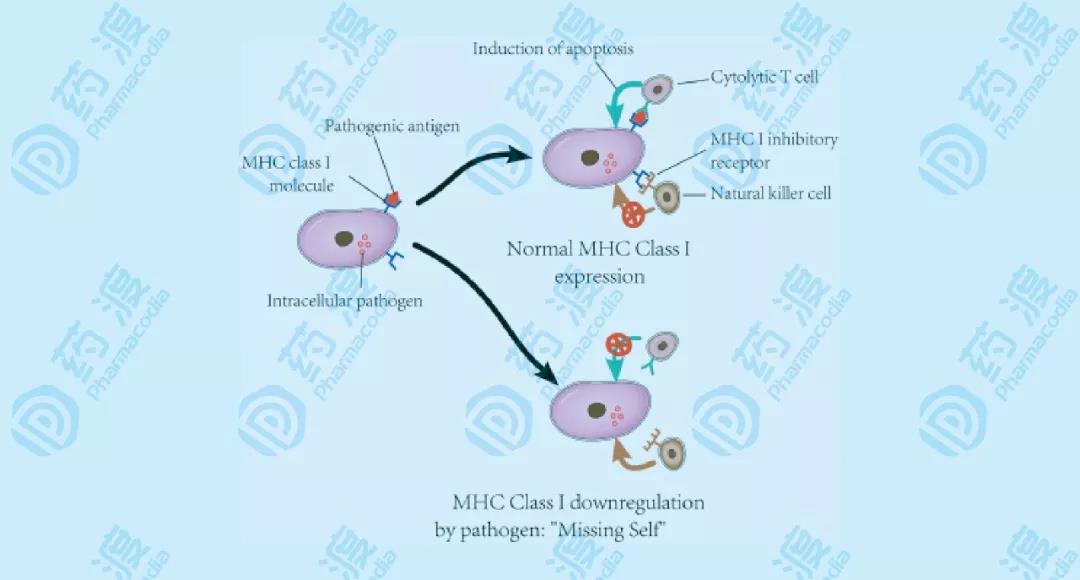
NK细胞,即自然杀伤细胞,是一种具有细胞毒性的淋巴细胞,属于天然免疫系统。NK细胞与获得性免疫系统中的细胞毒性T细胞,扮演着相近的角色。NK细胞对病毒感染的细胞,或者肿瘤形成,有着极快的响应速率。通常情况下,免疫细胞检测到感染细胞表面的MHC,引起细胞因子的释放,进而导致靶细胞裂解或凋亡。但NK细胞有所不同,它们可以在没有抗体或MHC的情况下,识别这些细胞并进行快速的免疫响应。对于那些失去自身标记的MHC-I型的细胞,NK细胞不经过激活就可以进行杀伤。而这些细胞通常是有害的,不能被其他免疫细胞发现并消灭,比如细胞毒性T细胞。(图2)
图2 NK细胞的“丢失自我”的杀伤机制 NK细胞通过自身表面的激活型和抑制型受体,调节自身的细胞毒性,比如杀伤细胞类免疫球蛋白受体。大部分受体不仅表达在NK细胞上,也表达在T细胞上。抑制型受体识别并结合MHC-I,这样也可以解释NK细胞杀伤那些没有表达MHC-I的细胞。而MHC-I在抗原呈递过程中,激活细胞毒性T细胞。但是,当被感染或变异的细胞,逐渐降低表达MHC-I,使它们自身免于被T细胞发现,规避T细胞免疫。而NK细胞正好弥补了这一点。
尽管NK细胞并不需要肿瘤相关抗原识别,来调整抗肿瘤的响应,但NK细胞同样存在免疫检查点的激活或抑制机制。虽然目前在细胞因子治疗方法,以及NK细胞过继转移等方面有所进步,但是,肿瘤细胞表达的针对NK细胞免疫检查点的配体,仍然能够抑制NK细胞介导的肿瘤细胞裂解。于是NK细胞功能缺失,肿瘤逃逸,病情加剧。因此目前有一些新的药物被研发出来,针对肿瘤-NK细胞的抑制型免疫检查点,以限制这种抑制作用。
当细胞在病毒感染向肿瘤转化期间,产生了应激压力或DNA损伤,就会产生胚系编码配体(germ-line ligand),这些配体可以被NK细胞表达的胚系编码受体(germ-line receptor)识别。而当细胞出现低表达MHC-I的情况时,就会触发“丢失自我”杀伤机制。因此为了最大程度地减少对正常细胞或组织的杀伤,必须微妙的平衡这种激活或抑制的信号,以调节NK细胞的活性。
NK细胞表面激活型的受体包括:自然细胞毒性引发受体(NCRs)、SLAM家族受体、c型凝集素、CD16(FcγRIII)。例如CD16并不识别细胞表达的配体,而是识别细胞结合的IgG抗体的Fc部分,而且单独通过CD16就足够引发强大的激活信号且克服大部分抑制信号,引发NK细胞-抗体介导的ADCC。另外,C型凝集素的同二聚体NKG2D,识别细胞表面因DNA损伤或应激压力而上调的分子。这些激活型受体结合配体后,还能引发细胞因子比如IFN-γ、TNF-α的分泌,其中IFN-γ可诱导周围细胞MHC-I的表达,增强CD8+ T细胞的识别能力(图1)。
NK细胞表面抑制型的受体包括:杀伤细胞免疫球蛋白类似受体(KIRs)、c型凝集素受体(NKG2A/CD94)、白细胞免疫球蛋白类似受体(LILRs)、常见的免疫检查点受体(PD-1、TIM-3、LAG-3、TIGIT)。它们当中大部分的配体,是MHC-I,而广泛表达的MHC-I配体介导的抑制信号,对于NK细胞响应调节至关重要。这些抑制型受体的表达,因NK细胞亚群不同而不同,比如CD56bright的NK细胞都表达NKG2A/CD94,而不表达KIRs,但是CD56dim的细胞只有约50~60%表达NKG2A/CD94,70~75%表达KIRs。
图3 NK细胞与肿瘤细胞间的激活或抑制型受体-配体相互作用 NK细胞的响应被这些激活或抑制型的相互作用的平衡微妙地调节,而这些NK细胞受体的表达取决于NK细胞的亚群,以及肿瘤微环境中的细胞因子或可溶性配体。同时,肿瘤细胞表达对应的配体,也依赖于肿瘤类型和微环境。
肿瘤细胞分裂期间,一定程度地引起DNA损伤,这会诱导NKG2D和DNAM-1的表达,进而引发NK细胞对肿瘤细胞的杀伤。然而,肿瘤细胞可以通过上调非经典的MHC-I,即HLA-G的表达,结合NK细胞的抑制型受体LIR-1,规避NK细胞的识别和杀伤。同时,肿瘤细胞也可以通过可溶性的NKG2D的配体,规避杀伤。这些配体通过可变剪切的方式,从肿瘤细胞表面脱落。于是NK细胞难以通过激活型受体NKG2D与其配体结合,也就难以激活对肿瘤细胞的杀伤。同时,这些可溶性的NKG2D配体,可以结合远处近处的NK细胞的NKG2D激活型受体,使其处于持续激活状态,而降低了NK细胞识别的敏感性。 一些位于肿瘤微环境中的抑制型免疫细胞,比如骨髓衍生抑制细胞(MDSCs)、调节型T细胞(Treg)可以抑制NK细胞的抗肿瘤活性。MDSCs通过分泌抑制型细胞因子IL-10和TGF-β,其中TGF-β可以下调NK细胞表面NKG2D的表达,或者通过细胞接触的方式,抑制NK细胞的活性。同样的,Treg也可以通过膜表面的TGF-β抑制NK细胞的活性,且Treg也通过竞争性消耗IL-2以减少IL-2对NK细胞的激活。
关于NK细胞的免疫检查点,同样存在着可能的负调控机制(图3展示了一部分): PD-1,在B细胞和T细胞表面存在诱导性表达,同样的,在NK细胞表面也有表达,尽管表达特性并不清楚,但PD-1减弱免疫功能的机制是明确的。不过,当NK细胞提升对肿瘤细胞的响应时,特别是IFN-γ分泌时,可能会导致肿瘤细胞PD-1配体的上调表达,从而反馈抑制NK细胞的响应。
CTLA-4,在激活的鼠源NK细胞上发现存在表达。但目前几乎没有线索能直接说明,人源NK细胞表达CTLA-4的相关活性。
TIGIT,带有Ig和ITIM(细胞内基于酪氨酸的抑制模体)结构域的T细胞免疫受体,通常在NK细胞上有所表达,属于抑制型受体,与DNAM-1共享PVR和Nectin-2受体。许多肿瘤过表达TIGIT的配体,CD155,这与肿瘤的增殖和迁移有关。在肿瘤环境中,CD8+T细胞和Treg都会上调表达TIGIT,而阻断TIGIT能够增强T细胞的功能。类似的,阻断TIGIT能够增强NK细胞分泌细胞因子和细胞毒性的能力。有数据表明,MDSC通过TIGIT信号通路,抑制NK细胞的活性。
KIR,杀手细胞免疫球类似受体,有抑制型和激活型两种,针对抑制型的KIR的阻断,是免疫治疗的主攻方向。抑制型KIR有两类,表达2个胞外免疫球类似结构域(KIR2DL),或者表达3个免疫球类似结构域(KIR3DL)。两类KIR的信号通路都通过ITIM(细胞内基于酪氨酸的抑制模体)实现。KIR能够识别并结合MHC-I,以抑制NK细胞的活性。在NK细胞的发育和稳态阶段,KIR与自身的MHC-I的相互作用,对于NK细胞“教育”的动态过程非常关键。尽管在肿瘤环境中,NK细胞上调表达激活型受体,但许多肿瘤能够保留它们的MHC-I,从而能够限制KIR表达NK细胞的响应和杀伤能力。而KIR信号通路的阻断型抗体,能够起到一定的肿瘤治疗的效果(图4)。
KIR抗体,IPH2101,在针对那些完全缓解状态的急性髓细胞样白血病患者的I期临床研究中,显示KIR结合发生在90%以上的NK细胞中(2周,最小剂量1mg/kg体重)。KIR抗体的治疗也升高了TNF-α和MIP-1β的血清浓度,以及NK细胞早期的激活标签CD69。在多发性骨髓瘤的治疗中,KIR抗体也产生了类似的效果。然而,在关于郁积性多发性骨髓瘤(MM)的II期临床中,并没有显著疗效。这可能与IPH2101介导的KIR2D受体的胞啃作用有关,即通过KIR抗体的ADCC作用,KIR2D受体被“啃掉”并转移到其他免疫细胞。尽管KIR抗体有效地阻断了KIR2D的信号通路,但是也阻断了这些表达KIR2D的NK细胞被“教育”(结合MHC-I)的能力,最终可能导致NK细胞对MM细胞的响应清零。这个难题也揭示了,在复杂的生物系统中,靶向检查点抑制研发所存在的一些挑战。
图4 NK细胞相关免疫检查点的抗体,以及它们的临床研究进展(截至2017年) C型凝集素异二聚体NKG2A/CD94,在NK细胞和CD8+T细胞上都有表达。它属于抑制型受体,对应的配体是HLA-E,可强烈抑制血液循环中的NK细胞。在很多肿瘤类型比如实体瘤或血癌中,上调编导HLA-E,从而减弱表达NKG2A的NK细胞的响应。在异源和自体造血干细胞移植中,NKG2A广泛表达于新生的NK细胞上,它与HLA-E的相互作用成为移植性治疗后NK细胞活性的主要抑制因素。在这个条件下,NK细胞通过减少NKG2A的表达,恢复NK细胞功能,并最终成熟。但在NK细胞完全成熟之前,阻断NKG2A也可以恢复功能,因此NKG2A的功能抑制型抗体也有希望用于治疗肿瘤。 Tim-3,T细胞免疫球和黏液素结构域包含分子,是T细胞调节免疫响应的负调节因子。小鼠中抗Tim-3的作用,导致自发性的自身免疫作用。在晚期胃癌和肺腺癌患者的外周NK细胞中,Tim-3上调表达。同时,在75%的胃肠道间质瘤患者中,肿瘤滤过性的NK细胞上有表达Tim-3。NK细胞表达的Tim-3的具体功能并不明确。源自晚期黑色素瘤的患者的功能受限的NK细胞,通过Tim-3拮抗剂的治疗,功能可获得恢复。然而,阻断Tim-3和它的配体galectin-9的相互作用,减少了健康NK细胞对急性髓性白血病(AML)的IFN-γ的产生。在对PD-1的阻断有抗性的患者中,Tim-3发生了阻断,同时Tim-3表达被上调,因此Tim-3被认为存在抑制免疫活性的作用。 Lag-3,淋巴细胞激活基因3,表达在CD4+和CD8+T细胞上。因只有小部分NK细胞表达Lag-3,并且NK细胞与MHC-II并不相互作用,因此Lag-3的功能还不是很清楚。
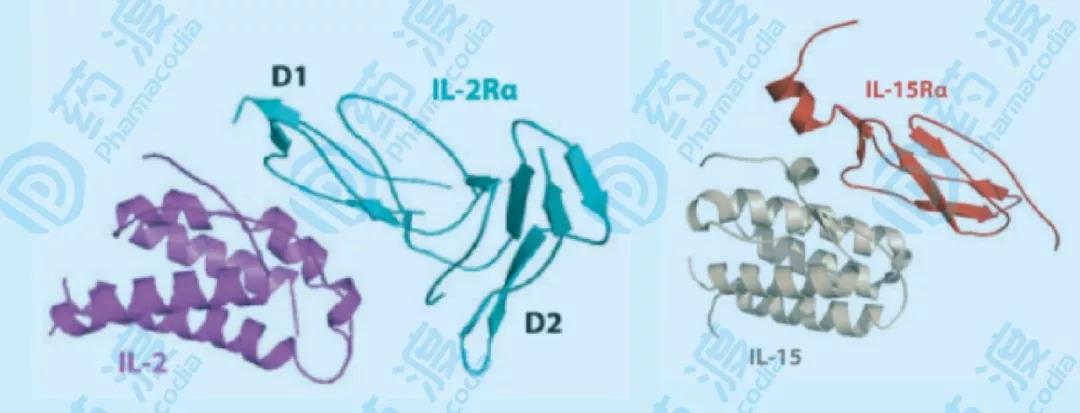
图5 IL-2/IL-2Rα和IL-15/IL-15Rα的结构示意图 IL-2和IL-15共享受体的两个亚基,即IL-2Rβγ和IL-15Rβγ完全相同,双亚基复合体以中亲和力受体存在,而结合了IL-2Rα或IL-15Rα的三亚基复合体,则以高亲和力受体存在。NK细胞表达中等亲和力的IL-2和IL-15受体。通常情况下,IL-2通过顺式作用,即先于同一细胞的IL-2Rα结合,然后与βγ结合,但NK细胞没有表达IL-2Rα,需要较高浓度的IL-2才能激活。而IL-15通过反式作用,即先于另一细胞(巨噬细胞、DC细胞)的IL-15Rα结合,然后与βγ结合。
NK细胞可以通过两种方式克服抑制,一是激活型细胞因子,比如IL-2、IL-15,二是CD16(FcγRIII)介导的NK细胞激活。IL-2和IL-15共享相同的βγ-受体亚基,而各自的α受体亚基可以提高它们在受体上的亲和力,它们结合对应的高亲和或中亲和受体,激活JAK-STAT信号通路,最终诱导更多的细胞因子的表达,细胞毒性的效应功能,以及增殖和存活。
IL-2治疗已经被大量研究,但是单独使用IL-2所产生的疗效并不显著。IL-2的一项缺点是,尽管它能够激活NK细胞,但同时,它也可以增强Treg的活性,限制NK细胞的响应。高剂量IL-2治疗肾癌或转移性黑色素瘤,只能让小部分患者的病情缓和,并且残留有大量细胞毒性。因为低剂量的IL-2更利于激活Treg的功能,所以IL-2治疗受限,而IL-15对于NK细胞依然有较好的刺激效果,且不会激活Treg的功能。
IL-15被用于实体瘤的治疗,以及在白血病患者中,维持NK细胞数量和活性。临床前、非人类哺乳动物以及早期临床的数据都表明,IL-15可以诱导NK细胞数量上升。其中IL-15与IL-15Rα的反式呈递,对最大化IL-15功效是必要的。ALT-803(IL-15N72D/IL-15Rα-Fc超级激动剂),在最近针对卵巢癌的鼠模型研究中,使NK细胞非常显著地去颗粒化,并导致细胞因子的大量产生。如ALT803能够使卵巢癌患者腹水中的NK细胞恢复活性。同时,在抗CD20抗体的联合作用下,ALT803增强了CD16引发NK细胞对B淋巴细胞的清除。IL-15一方面能够协助越过免疫检查点的抑制机制,另一方面能够完善NK细胞上CD16介导的功能,因此它成为了新的热门免疫治疗靶点。
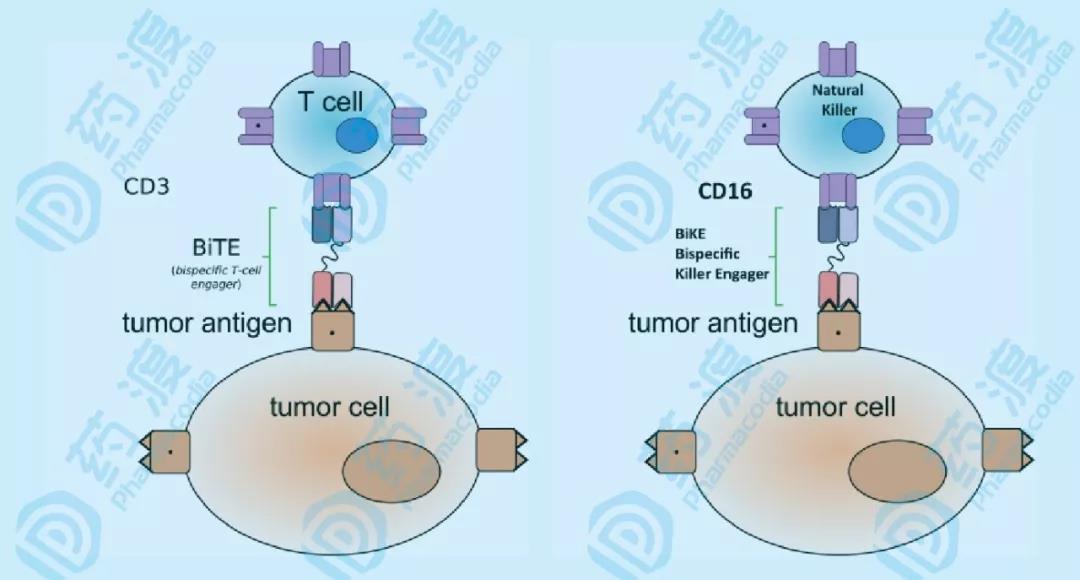
CD16a,即FcγRIIIa,NK细胞表达的Fc低亲和受体,介导抗体的直接杀伤ADCC。CD16a表达在CD56dim的NK细胞上,这些NK细胞在健康个体内,占有至少80%的所有外周的NK细胞。CD16a与Fc结合后,通过ITAM信号通路,导致细胞因子的产生和细胞的去颗粒化。与NK细胞的其他激活型受体不同,CD16a与Fc的结合不需要协同激活,就可以产生强力的响应,这也使得NK细胞能够在病毒感染和肿瘤形成早期,通过抗体产生免疫反应。然而,NK细胞表达的CD16a对不同抗体的Fc亲和力有所差异,介导的ADCC的效果参差不齐。因此,开发BiKE和TriKE之类的分子,能改进亲和力,并且可以针对不同的肿瘤相关抗原。
图6 BiTE和BiKE BiTE能够同时结合T细胞和肿瘤细胞,具有两条scFv串联结构的抗体,分别结合T细胞的CD3ε(TCR亚基)和肿瘤细胞的肿瘤相关抗原。而BiKE则能够同时结合NK细胞和肿瘤细胞,结构与BiTE相近,结合NK细胞的CD16a,强化ADCC作用,以及肿瘤相关抗原。新一代的BiTE甚至加入了抗PD-1/PD-L1的scFv,以减少免疫检查点抑制。类似的原理(图3),BiKE可以融合IL-15或拮抗前述受体信号通路的scFv(TriKE),移除免疫抑制或增强免疫响应。
激活NK细胞、解除NK细胞的免疫抑制、限制NK细胞毒性的时空。 新的针对NK细胞的研究,比如仿照BiTE思路而来的BiKE(图6),以及仿照CAR-T思路的CAR-NK(另文描述)。BiTE加上抗PD-1的部分成为了CiTE(检查点抑制T细胞连接抗体),那么BiKE加上了IL-15的部分而成为了TriKE(三功能NK细胞连接抗体,图7,8):
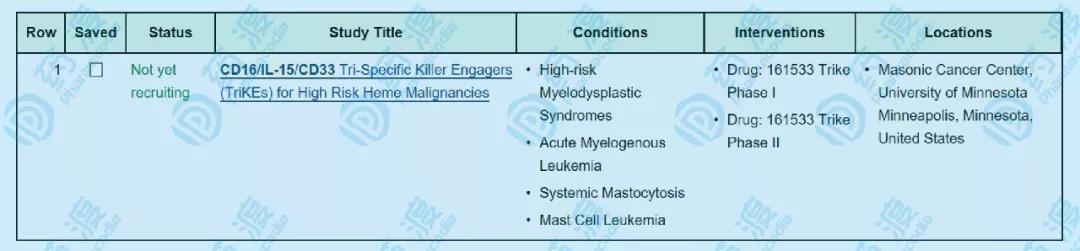
图7 CiTE和TriKE 图8 TriKE的临床研究正在招募中
如果说,TriKE给我们提供了增强NK细胞的免疫响应和特异性的参考,那么如何武装NK细胞,是可以关注的点。一方面,抑制NK细胞的抑制性信号通路,比如抗体阻断NK细胞表面的抑制型受体,或者激活NK细胞,通过结合激活型受体的配体,比如IL-2、IL-15、Fc等;另一方面,提高NK细胞,尤其是渗透到肿瘤组织的NK细胞的有效性,提高针对肿瘤细胞的特异性,比如利用抗肿瘤的单抗。那又如何将这几方面的优势综合起来“武装”NK细胞?
细胞因子对肿瘤的治疗效果,因为受到药物传递的限制,无法在肿瘤病灶部位产生足够的活性,比如之前提到过IL-2的局限性。为了最大化细胞因子的治疗效果,重组的抗体-细胞因子融合蛋白被广泛研究,以增强单抗靶向肿瘤的能力。如此,细胞因子通过单抗,被引导至特异性的肿瘤部位,能够刺激引发更多有效的抗肿瘤响应,并且避免单独使用细胞因子时会造成的系统性的细胞毒性。
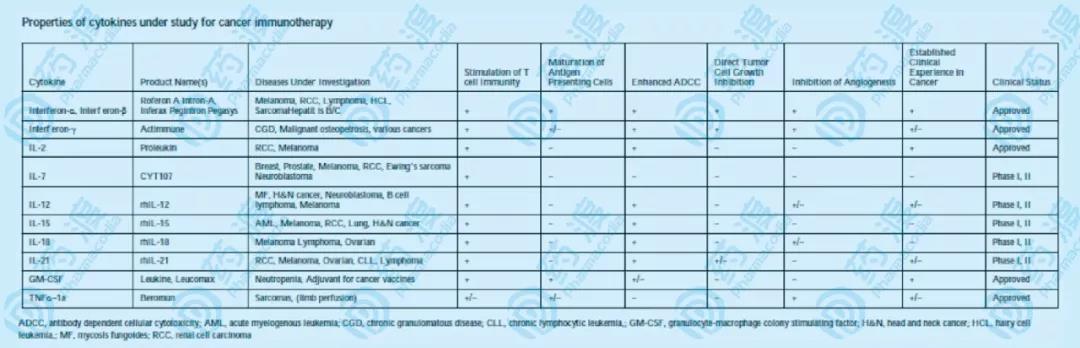
图9 用于免疫治疗的细胞因子,其中一些还处于临床研究阶段 细胞因子是多肽或蛋白质,在重组基因表达上,容易实现在哺乳动物细胞中的表达,只需将INFs、ILs之类的融合在抗体的N端或C端。比如罗氏在研的RG7813(图10),包含抗CEA单抗,且其中一条重链的C端融合改造后的IL-2。同样的,前文提到的TriKE,基于scFv抗体,以拉近NK细胞和肿瘤细胞,并通过融合细胞因子激活NK细胞的活性。
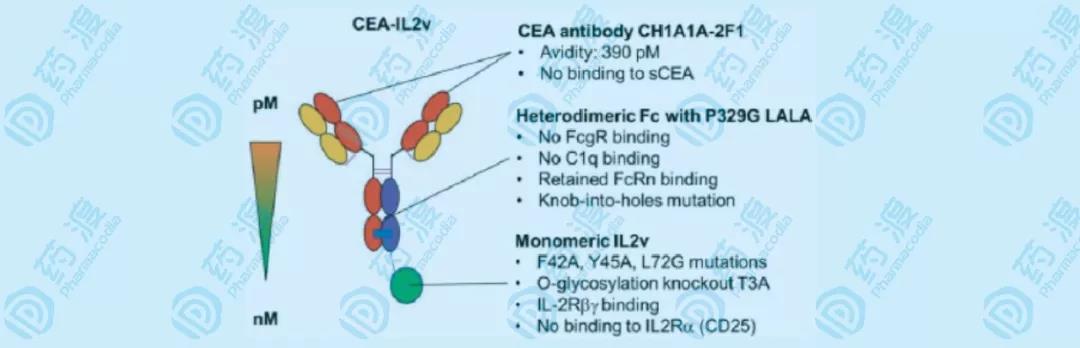
图10 RG7813示意图 抗CEA单抗特异性结合CEA最靠近细胞膜表面的结构域,而不结合因酶切作用而进入血液的可溶性部分,sCEA属于肿瘤细胞抑制体液免疫的作用。IL-2通过突变改造,偏向于结合IL-2中等亲和力受体IL-2Rβγ,从而激活NK细胞或CD8+细胞毒性T细胞。因为IL-2存在,Fc的ADCC/CDC的活性被取消,为了长效作用仍保持FcRn的结合,并且因为单侧结合IL-2,引入KiH技术实现Fc的异二聚化。 设计理想中的抗体-细胞因子融合蛋白,包括以下步骤: 选择合适的靶向抗原:在正常组织中表达量极低的肿瘤相关抗原,以减少抗体“沉没”在正常组织中而无法发挥药效,同时也需要肿瘤组织中尽可能高表达,以便富集足够多的细胞因子。细胞表面的抗原,结合后不会被细胞吸收,这样可以延长融合细胞因子的半衰期。肿瘤微环境相关抗原也可以,比如与肿瘤迁移、浸润相关的细胞因子。在血液循环中没有显著的数量,比如sCEA,能够持续性消耗激活的免疫细胞,使这些免疫细胞无法造成有效杀伤。
选择合适的细胞因子:考虑细胞因子靶向到肿瘤细胞的目的,还没有一种单一的细胞因子具有所有的抗肿瘤的特性。是为了激活和辅助增殖T细胞、NK细胞还是巨噬细胞?是否需要直接作用于肿瘤细胞,还是用于抑制肿瘤组织血管的形成?细胞因子介导的杀伤机制,诱导T细胞的细胞毒性,还是ADCC,还是直接作用?本文优先考虑对NK细胞的激活作用。
结构的选择:完整的抗体结构,scFv,Fab等等。
融合细胞因子的生物活性:通过定量比较,维持完全的亲和力和生物活性,或者减弱活性以减少脱靶造成的系统性细胞毒性。正常组织中,过高的亲和力可能导致细胞毒性。而结合到靶细胞后,也要注意抗体介导的巨噬细胞吞噬,除非细胞因子靶向作用于巨噬细胞。这会使融合的细胞因子被巨噬细胞吞噬和消除。因此,如果用到了Fc融合,Fc往往需要通过改造以减少细胞因子的非特异消耗。
PK和PD:足够长的半衰期,以便于在肿瘤部位富集。
临床前药效估计:在肿瘤模型动物实验中,检测抗肿瘤药效和免疫毒性。
剂量和使用规划:在肿瘤微环境以及宿主的免疫效应细胞的条件下,基于耐受性和药代的效果制定。
结 尾 抗体细胞因子的融合,要求它针对肿瘤细胞的活性和有效性,强于单独用抗体或单独用细胞因子的效果,否则联合用药可以达成更好的效果。因此,最好在相关同基因的免疫活性动物模型中,验证融合蛋白的效果。 最后,以TriKE的构建理念为结尾,一方面靶向肿瘤细胞,一方面靶向NK细胞,激活NK细胞或者解除NK细胞的免疫抑制,然后通过IL-2或IL-15的改造体激活NK细胞的活性。如果融合的细胞因子,在TriKE功能的基础上,活性可限制在肿瘤微环境中释放,而且本身在血液循环中有较长的半衰期,这可能比TriKE更为理想。
1. NLRC5/MHC class I transactivator is a target for immune evasion in cancer.(2016), PNAS, Sayuri Yoshihama, Jason Roszik, Isaac Downs, et al. 2. Trends in the global immuno-oncology landscape. (2018) Nature Reviews. Jun Tang, Laura Pearce, Jill O’Donnell-Tormey and Vanessa M.Hubbard-Lucey. 3. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. (2018) Cell. Miguel F. Sanmamed and Lieping Chen. 4. The basic principles of chimeric antigen receptor (CAR) design. (2013) Cancer discovery. Michel Sadelain, Renier Brentjens, and Isabelle Riviere 5. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. (2017) Oncoimmunology.Christian Klein, Inja Waldhauer, et al. 6. Targeted killing of colorectal cancer cell lines by a humanised IgG1 monoclonal antibody that binds to membrane-bound carcinoembryonic antigen.(2008) British Journal of Cancer.PJ Conaghan, SQ Ashraf. 7. Natural Killer Cells Unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. (2017) SeminImmunology. Zachary B. Davis, Daniel A. Vallera, Jeffrey S. Miller, and Martin Felices. 8. Antibody-cytokine Fusion Proteins for Treatment of Cancer: Engineering Cytokines for Improved Efficacy and Safety. (2014) Semin Immunology. PatriciaA. Young, Sherie L. Morrison, and John M. Timmerman. 9. ESMO HANDBOOK OF IMMUNO-ONCOLOGY. Edited by John B.A.G. (2018) Haanen,Raffaele Califano, Iwona Lugowska, Marina Chiara Garassino.
康宁/Corning® 淋巴细胞无血清培养基
|
购买进口仪器、试剂和耗材——就在始于2001年的毕特博生物
www.bitebo.com |
|